
Was ist Phosphatdiabetes?
Letzte Aktualisierung: 17.07.2020
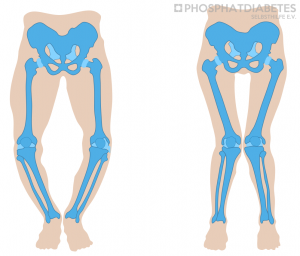
Die X-chromosomal-dominante hypophosphatämische Rachitis ist eine angeborene Störung, die neben dem Skelettsystem u.a. auch die Zähne in ihrer Mineralisation beeinflusst.
Die Erkrankung kann ab dem Ende des ersten Lebensjahres, meist mit dem eigentlichen Laufalter im zweiten Lebensalter, durch einen watschelnden, breitbeinigen Gang und einem verminderten Längenwachstum auffallen. Bei einzelnen Betroffenen kann die Diagnose auch erst im Schulalter aufgrund von Beinschmerzen und / oder einer zunehmenden Beinachsenfehlstellung (O- oder X Beine) diagnostiziert werden. Ist ein Elternteil und gar beide von der XLH betroffen, kann die Diagnose möglicherweise schon in den ersten Lebensmonaten diagnostiziert werden.
Ursache der Erkrankung
Ein gesunder fester Knochen besteht aus einer Knochengrundsubstanz sowie aus einem bestimmten Verhältnis der Mineralien Calcium und Phosphat. Kommt es nun zu einer Störung einer dieser drei wichtigen Bestandteile, so kann es zu einer Weichheit des Knochens und damit besonders an den unteren Extremitäten zu einer Verbiegung der Ober- und/oder Unterschenkel oder auch zu einer Fraktur kommen.
Bei der oben genannten Form der hypophosphatämischen Rachitis (XLH) kommt es durch eine genetische Veränderung im sogenannten PHEX-Gen zur vermehrten Bildung des Hormons Fibroblast Growth Factor 23 (FGF23). FGF23 wird in den Knochenzellen gebildet und reguliert normalerweise neben dem Vitamin D den Phosphat-Haushalt des Körpers. Kommt es nun aber durch die genetische Veränderung zu einer vermehrten Bildung von FGF23, so führt dies zu einer vermehrten Ausscheidung von Phosphat über die Nieren. Normalerweise wird 90% des von der Niere filtrierten Phosphats in den Körper zurückgeführt. Durch die gesteigerte FGF23-Konzentration kommt es aber zu einer Störung dieser Rückresorption und zu einer überproportional starken Ausscheidung von Phosphat über die Nieren in den Urin. Dieses führt im Blut zu einem niedrigen Phosphat Spiegel und damit zu einer verminderten Konzentration von Phosphat im Skelettsystem. Darüber hinaus vermindert FGF23 die Synthese von aktivem Vitamin D in den Nieren. Durch den Phosphat-Mangel im Knochen und Mangel an aktivem Vitamin D kommt es zu einer Mineralisationsstörung und damit zu einer Weichheit des Knochens, die sich besonders in den Beinen darstellt und als hypophosphatämische (= zu wenig Phosphat) Rachitis bezeichnet wird.
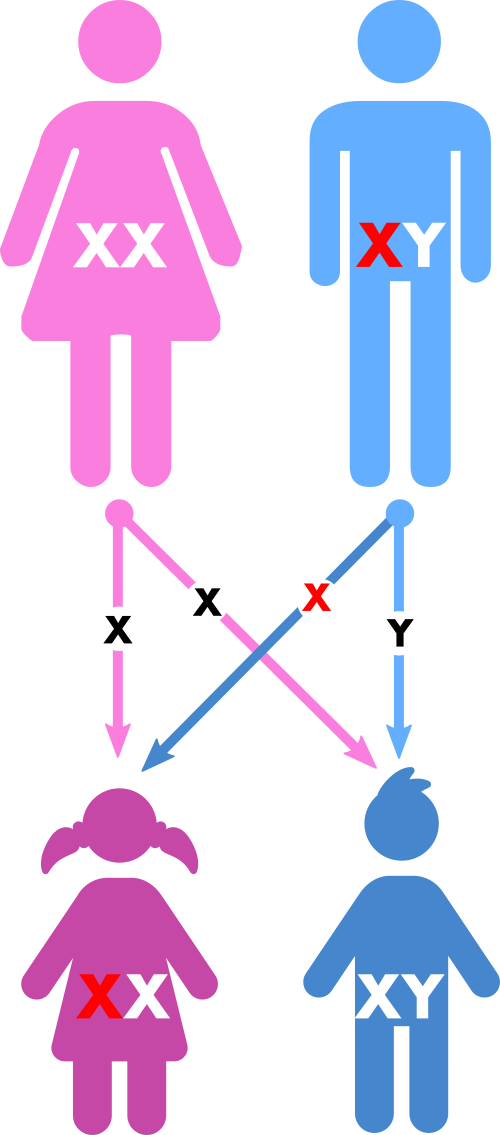
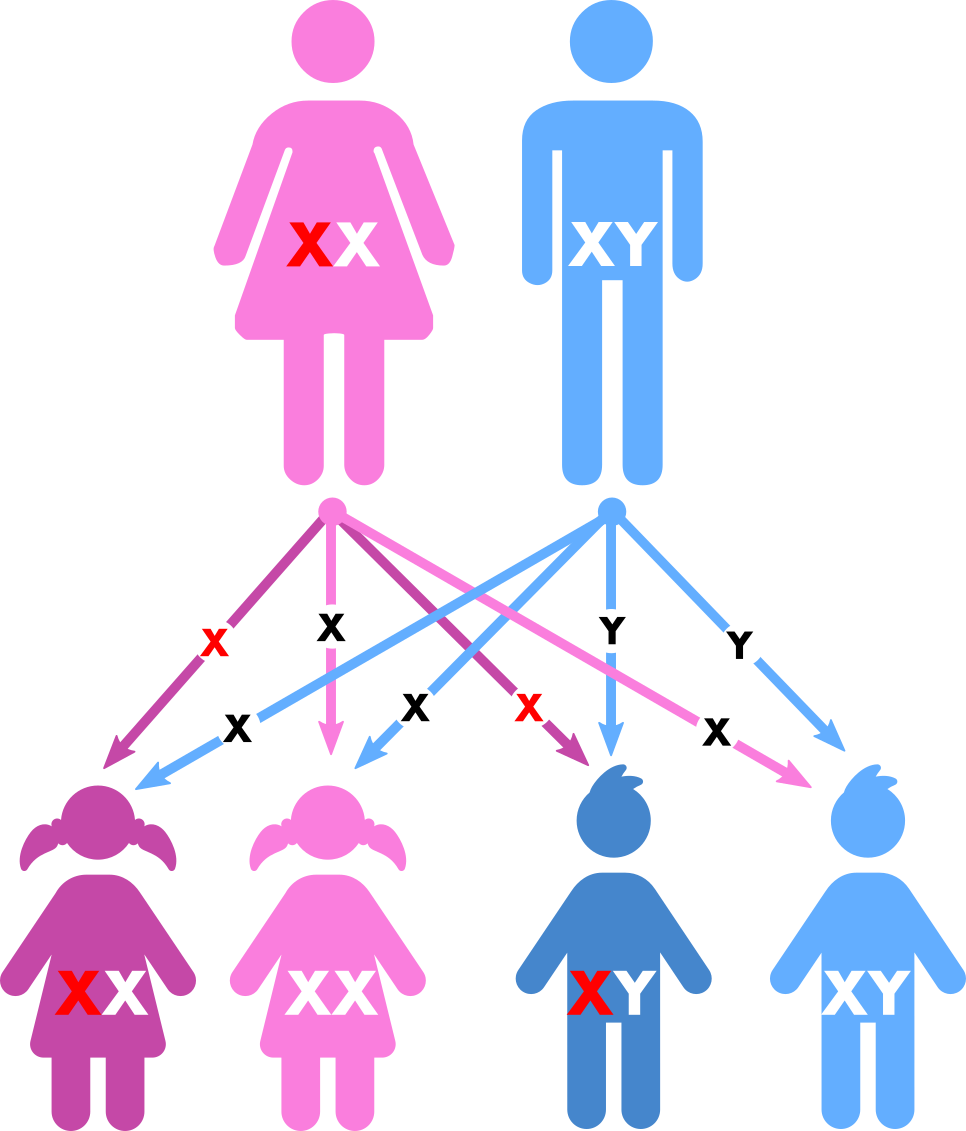
Die Erkrankung ist an das X-Chromosom gekoppelt. Das heißt, bei einer Erkrankung des Vaters sind alle Töchter betroffen und alle Söhne gesund, während statistisch die Hälfte der Söhne und die Hälfte der Töchter einer an Phosphatdiabetes erkrankten Mutter betroffen sein können. Die Ausprägung der Erkrankung ist durchaus auch innerhalb einer Familie sehr verschieden.
Mittlerweile sind über 300 verschiedene krankheitsauslösende genetische Veränderungen im PHEX-Gen (Mutationen) beschrieben. Neben der vererbten Form eines Phosphatdiabetes gibt es Patient*innen aus Familien, in denen zuvor weder Vater noch Mutter an einem Phosphatdiabetes erkrankt waren. Dies ist etwa bei einem Drittel der Patient*innen der Fall. Man spricht dann von einer Spontan-Mutation.
 |
 |
|
| Vererbungsmodus des Phosphatdiabetes anhand des Stammbaums bei einem an Phosphatdiabetes erkrankten Vater. | Vererbungsmodus des Phosphatdiabetes anhand des Stammbaums bei einer an Phosphatdiabetes erkrankten Mutter. |
Die Häufigkeit dieser seltenen Knochenerkrankung wird auf etwa ein betroffenes Neugeborenes auf 20.000 bis 25.000 Neugeborene beziffert. Trotz dieser Seltenheit ist es die häufigste angeborene Rachitis-Form.
Diagnose
Kommt es nach dem Beginn des freien Laufens im zweiten Lebensjahr zu einer zunehmenden Verbiegung der unteren Beine (X- oder O-Beine) oder möchte ein Kleinkind am Ende des zweiten Lebensjahres nicht gerne laufen, sollte der der/die Kinderarzt*Kinderärztin eine Blutuntersuchung durchführen. In dieser Untersuchung ist zunächst ein Vitamin D-Mangel auszuschließen. Dieser kommt sehr viel häufiger als ein Phosphatdiabetes vor und kann ebenfalls Ursache einer Deformierung der Beine im Kleinkindesalter sein. Haben aber die Eltern die Vitamin D-Prophylaxe regelmäßig im ersten und zweiten Lebensjahr durchgeführt, so ist ein Vitamin D Mangel als Ursache der Beinverformung sehr unwahrscheinlich. Zeigt sich bei der Blutabnahme ein erniedrigter Serum-Phosphat-Wert (<1 mmol/l) und ist der Marker für den Knochenumsatz erhöht (Alkalische Phosphatase), dann sollte eine Vorstellung des Kindes beim Spezialisten/Spezialistin für Hormonstörungen (Kinderendokrinologie: www.dgked.de) oder beim Spezialisten/ Spezialistin für Nierenerkrankungen (Kindernephrologie: www.gpn.de) erfolgen.
Diese führen dann weitere Laboruntersuchungen durch, um die Verdachtsdiagnose Phosphatdiabetes zu bestätigen oder zu entkräften. Neben zusätzlichen Laborwerten ist auch eine Röntgenaufnahme zumeist der linken Hand erforderlich. In dieser Röntgenaufnahme könnte man dann bei Vorliegen eines Phosphatdiabetes sogenannte rachitische Veränderungen finden. Diese sind Hinweise auf eine gestörte Mineralisation des Knochens. Die Erkrankung kann bei ca. 80% der Patient*innen durch eine genetische Untersuchung weiter gesichert werden. Dies sollte bei negativer Familienanamnese oder unklaren Befunden angestrebt werden.
Bei Säuglingen von Erkrankten kann die Diagnose noch vor Manifestation von Symptomen etwa ab dem 6. Lebensmonat laborchemisch und/oder auch bei bekannter Mutation bereits in den ersten Lebensmonaten genetisch gestellt werden.
Therapie
Die Behandlung bei dieser Form der hypophosphatämischen Rachitis erfolgt entweder mit der Substitutionstherapie (Ersatz von Phosphat und Vitamin D) bestehend aus mindestens 4 bis 6 über den Tag verteilten Gaben von Phosphat (zumeist als Lösung, bei älteren Kindern auch mittels Phosphatkapseln) in Kombination mit aktivem Vitamin D (Wirkstoff: Alfacalcidol oder Calcitriol) als Tropfen bzw. Tabletten. Es gibt alternativ dazu eine Antikörper-Therapie, die ab dem 2. Lebensjahr mit dem zugelassenen Medikament Burosumab (siehe unten) erfolgt. Mit der Kombinationstherapie aus aktivem Vitamin D und Phosphat wird versucht, das über die Nahrung zugeführte Phosphat vermehrt über den Darm aufzunehmen und in den Körper einzuschleusen. Im Idealfall gelingt es, so viel Phosphat ins Skelettsystem zu transportieren, dass sich die Aktivität des Knochenumsatz-Wertes (Alkalische Phosphatase im Serum) weitestgehend normalisiert, der Knochen gut mineralisiert und damit stabil und belastbar wird und sich das Wachstum verbessert. Je früher die Therapie eingeleitet wird, umso geringer ist das Risiko der Entstehung verstärkter Beinachsenfehlstellungen. Leider kann es bei der Einnahme insbesondere des Phosphats zu Problemen kommen, denn das
Phosphat schmeckt manchen Kindern nicht. Größere Gaben von Phosphat können zu Bauchschmerzen und Durchfall führen. Deshalb ist es wichtig, die tägliche Phosphatmenge auf möglichst viele kleine Portionen zu verteilen. Seit Frühjahr 2018 steht für Kinder mit Phosphatdiabetes ab dem 2. Lebensjahr mit dem röntgenologischen Nachweis einer Knochenerkrankung eine zugelassene Therapie mit einem Antikörper zur Verfügung, der das erhöhte körpereigene FGF23 gezielt bindet und neutralisiert. Das Medikament mit dem Wirkstoff Burosumab wird alle 14 Tage unter die Haut gespritzt. Dies wird fachmännisch von einem Homecare Service durchgeführt. Sobald keine Dosisanpassung mehr zu erwarten ist, ist bei manchen Patient*innen eine Selbstinjektion/Injektion durch eine Betreuungsperson möglich. Voraussetzung ist die Einweisung in die Injektionstechnik durch medizinisches Fachpersonal. Die erste Selbstinjektion/Injektion durch eine Betreuungsperson sollte unter Aufsicht von medizinischem Fachpersonal durchgeführt werden.
Seit 2020 ist Burosumab auch für Erwachsene zugelassen. Das Injektionsintervall bei Erwachsenen beträgt 28 Tage. Unter der Therapie kommt es meist nach wenigen Injektionen zu einer Normalisierung der ursprünglich deutlich erhöhten Phosphatausscheidung über den Urin. Der Blut-Phosphatwert steigt in den unteren Normalwertbereich. Das Skelettsystem wird nun ausreichend mit Phosphat versorgt, die Skelettmineralisation verbessert sich zunehmend. Mittel- bis längerfristig zeigt sich im Röntgenbild eine deutliche Verbesserung der Knochenmineralisierung. Nach mehrjähriger Therapie sind auch Ausgradigungen der ursprünglichen Beinachsenfehlstellungen beobachtet worden. Nach Beginn der Therapie sind zunächst in den ersten Wochen kurzfristige Vorstellungen zur Kontrolle der Verträglichkeit, aber auch zur Dosisanpassung von Burosumab erforderlich. Dies wird mit Blut- und Urinkontrollen überprüft. Nicht selten ist eine Erhöhung der Medikamentendosis erforderlich. Die Medikation wird in der Regel gut vertragen, schwere Nebenwirkungen wurden bisher nicht berichtet. Über evtl. entstehende langfristige Nebenwirkungen liegen noch keine Erkenntnisse vor, dies gilt es weiter zu beobachten. Das Für und Wider der beiden Therapiemöglichkeiten ist ausführlich in einer europäischen Therapieempfehlung dargelegt worden (siehe unten).
Kontrolle der medikamentösen Einstellung
Nach Beginn der medikamentösen Therapie, unabhängig ob durch Phosphat- und Vitamin D-Gaben oder durch Injektion des Antikörpers, ist zunächst alle 4 bis 6 Wochen, später dann alle 3 Monate, eine Vorstellung des Kindes bzw. Jugendlichen bei einem Spezialisten/Spezialistin sinnvoll. Bei diesen Kontrollvorstellungen erfolgen dann Untersuchungen des Urins sowie des Blutes. Der/die Spezialist*in wird dann in Abhängigkeit von den Laborwerten möglicherweise eine Korrektur der bisherigen Medikation durchführen.
Empfohlene zusätzliche Untersuchungen:
Nieren:
Durch die medikamentöse Therapie mit Phosphat und aktiven Vitamin D kann es vereinzelt zu Kalkablagerungen in der Niere kommen (Nephrocalcinose). Das Entstehen einer Nephrokalzinose unter der Antikörper-Therapie wurde bisher nicht beobachtet. Es sollte etwa alle ein bis zwei Jahre eine Ultraschalluntersuchung der Nieren erfolgen.
Skelettsystem:
Bei Menschen mit Phosphatdiabetes sind zumeist die Ober- und/oder Unterschenkel in starker X- oder O-Beinstellung gebogen. Dadurch kommt es zu einem auffälligen Gangbild. Je früher die Diagnose gestellt und eine Behandlung begonnen wird, umso größer ist die Möglichkeit, dass sich diese Beinfehlstellung wieder unter alleiniger Medikation korrigiert. Sollte sich diese aber nach einer konsequent durchgeführten medikamentösen Therapie über 3 bis 4 Jahren nicht einstellen, so ist eine Wachstumslenkung mittels Epiphyseodese (z.B. sogenannter Eight-Plates) in Betracht zu ziehen. Bei älteren Kindern / Jugendlichen kann auch ein größerer chirurgischer Eingriff („Umstellungsosteotomie“) erforderlich sein.
Wachstum:
Kinder mit einem Phosphatdiabetes sind bei Geburt in der Regel normal groß und schwer. In den ersten beiden Lebensjahren kommt es durch eine verlangsamte Wachstumsgeschwindigkeit zu einem schlechteren Körperlängenwachstum. Viele Kinder liegen mit ihrer Körperlänge zu Beginn des 3. Lebensjahr im Bereich der 3. Perzentile. Ein weiterer „Verlust“ an Körperlänge stellt sich dann in der Regel nicht mehr ein. Auch bei guter medikamentöser Einstellung und geraden Beinen sind Patient*innen mit einem Phosphatdiabetes als Erwachsene meist nur grenzwertig normalwüchsig, können aber bei großen Eltern als Erwachsene durchaus normalwüchsig sein.
Zähne:
Menschen mit einem Phosphatdiabetes können auch eine Störung der Zahnschmelz- und Dentinentwicklung der Milchzähne, aber auch der bleibenden Zähne aufweisen. Nicht selten werden Fisteln, entzündliche Veränderungen der Zahnwurzelspitzen mit Vereiterungen (Abszesse) bei einem karies-freien Gebiss beobachtet. Um möglichen Zahnverlusten entgegenzuwirken, sind regelmäßige zahnärztliche Untersuchungen sinnvoll. In allererster Linie kommt es aber auf eine gründliche tägliche Zahnpflege und regelmäßige Prophylaxe Maßnahmen an.
Gehör:
Sehr selten kommt es bereits im Kindes- und Jugendalter zu einer Hörstörung im Innenohr. Sollten sich Hinweise auf eine Hörstörung ergeben, so ist eine Abklärung durch einen Hals-Nasen-Ohren-Arzt/Ärztin sinnvoll.
Kopfentwicklung:
Sehr selten kommt es im Säuglings- und Kleinkindesalter zu einer auffälligen Entwicklung des Schädels durch den vorzeitigen Verschluss der Schädelnähte (Kraniosynostose). Dann sollte mit dem Kinderarzt/Kinderärztin über eine Vorstellung beim Kinder- Neurochirurg*in gesprochen werden.
Europäische Therapieempfehlungen:
Im Dezember 2017 hat sich eine Gruppe von 20 Experten aus dem Bereich Kinderheilkunde, Nephrologie, Endokrinologie, Orthopädie, Zahnheilkunde und Neurochirurgie, sowie Vertreter*innen der Patientenorganisation Phosphatdiabetes e.V. aus Deutschland und der Patientenorganisation aus Frankreich in Berlin getroffen, um Therapieempfehlungen für diese seltene Erkrankung zu erarbeiten. Diese sind seit Juli 2019 in einer sehr renommierten Fachzeitschrift publiziert (Haffner et al Nat Rev Nephrol 2019) und können kostenlos als open access publication eingesehen werden (https://doi.org/10.1038/s41581-019-0152-5 ). Anfang 2025 wurde dann eine Überarbeitung dieser Empfehlungen veröffentlicht. (Haffner D, Schnabel D, Kirchhoff M et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nature reviews nephrology https://doi.org/10.1038/s41581-024-00926-x )
Patientenregister:
Um mehr über den Krankheitsverlauf und die Effektivität der Therapie des Phosphatdiabetes bei Kindern und Jugendlichen zu erfahren, haben sich Kinderendokrinolog*innen und Kindernephrolog*innen aus Deutschland zusammengeschlossen, um ein Patientenregister zu initiieren. Hierbei werden nach entsprechender Aufklärung und schriftlicher Einverständniserklärung der Eltern und Patient*innen die relevanten klinischen Daten der Patient*innen nach entsprechender Unkenntlichmachung der persönlichen Daten („Pseudonymisierung“) in eine Datenbank eingegeben. Unterdessen konnten bereits 40 Behandlungszentren mit aktuell 150 Patient*innen in das Register eingeschlossen werden. Die Teilnehmenden des Studienregisters treffen sich einmal pro Jahr. Im Rahmen des Registers sind auch weitere klinische Studien zur Behandlung der Erkrankung vorgesehen. Inzwischen gibt es auf europäischer Ebene ein Europäisches Register für seltene Knochen- und Stoffwechselerkrankungen (EuRREB). EuRREB ist ein Zusammenschluss von EuRRECa (Europäisches Register für seltene endokrine Erkrankungen) und EuRR-Bone (Europäisches Register für seltene Knochenerkrankungen und Mineralisierungsstörungen). Eurreb beinhaltet das e-Rec (E-Reporting) und das Kernregister. e-REC ist ein elektronisches Berichtsprogramm, um neue Verdachtsfälle und neue bestätigte Fälle von seltenen Knochenstoffwechselstörungen zu erfassen. Das Kernregister sammelt allgemeine und krankheitsspezifische Datensätze für eine breite Palette endokriner Knochenerkrankungen. Sie ermöglicht das Beobachten des Krankheitsverlaufs über einen längeren Zeitraum und kann zur Verbesserung der klinischen Versorgung und zur Forschung beitragen. Patient*innen können einen Zugang zum Kernregister erhalten, um sich die vom Arzt/Ärztin eingetragenen Daten anzeigen zu lassen und durch das Ausfüllen von Fragenbögen (z.B. zur Lebensqualität, zu Schmerzen, usw.) zum Kernregister beitragen. Für Phosphatdiabetes gibt es innerhalb des Kernregisters ein krankheitsspezifisches Modul, in dem Aspekte, wie Diagnose, Therapie, Verlauf, usw. zur Erkrankung dokumentiert werden. Dies kann dauerhaft zum besseren Verständnis der Erkrankung beitragen. Weiterführende Informationen sind unter https://eurreb.eu/registries/e-rec/ und https://eurreb.eu/registries/core-registry/ zu finden.
Resümee:
XLH ist eine seltene angeborene Erkrankung, die im Wesentlichen das Skelettsystem und die Zähne der Patient*innen betrifft. Eine frühzeitige Diagnose und Behandlung ist notwendig, um Komplikationen, insbesondere am Skelettsystem aber auch anderen Organen, zu verhindern. Durch die Entwicklung und Zulassung von Burosumab sind die Möglichkeiten der medikamentösen Therapie wesentlich erweitert worden. Erstmals sind bei den behandelten Patient*innen normale Phosphatkonzentrationen im Blut nachweisbar, die nach ca. 9 bis 12 Monaten zu einer deutlichen Besserung der Knochenmineralisierung führen. Wegen der Beteiligung verschiedener Organe bei der Erkrankung sollte möglichst die Betreuung durch ein multidisziplinäres Team in einer Spezialsprechstunde erfolgen.
Literatur:
Haffner D, Emma F, Seefried L, Högler W, Javaid KM, Bockenhauer D, Bacchetta J, Eastwood D, Biosse Duplan M, Schnabel D, Wicart P, Ariceta G, Levtchenko E, Harvengt P, Kirchhoff M, Gardiner O, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenický P, Rejnmark L, Linglart A. Clinical practice recommendations for the diagnosis and management of X linked hypophosphataemia. Nat Rev Nephrol. 2025 Jan 15. doi: 10.1038/s41581-024-00926-x. Epub ahead of print. Erratum in: Nat Rev Nephrol. 2025 Feb 4. doi: 10.1038/s41581-025 00939-0. PMID: 39814982.
Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, Wicart P, Bockenhauer D, Santos F, Levtchenko E, Harvengt P, Kirchhoff M, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenicky P, Rejnmark L, Linglart A. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019 Jul;15(7):435-455. doi: 10.1038/s41581-019-0152-5. PMID: 31068690; PMCID: PMC7136170.
Kamenický P, Briot K, Munns CF, Linglart A. X-linked hypophosphataemia. Lancet. 2024 Aug 31;404(10455):887-901. doi: 10.1016/S0140-6736(24)01305-9. Epub 2024 Aug 21. PMID: 39181153.
Haffner D, Leifheit-Nestler M, Grund A, Schnabel D. Rickets guidance: part I-diagnostic workup. Pediatr Nephrol. 2022 Sep;37(9):2013-2036. doi: 10.1007/s00467-021-05328-w. Epub 2021 Dec 15. PMID: 34910242; PMCID: PMC9307538.
Haffner D, Leifheit-Nestler M, Grund A, Schnabel D. Rickets guidance: part II-management. Pediatr Nephrol. 2022 Oct;37(10):2289-2302. doi: 10.1007/s00467-022-05505-5. Epub 2022 Mar 29. PMID: 35352187; PMCID: PMC9395459.
| Dr. med. Dirk Schnabel, Ltd. Oberarzt Stellv. Leiter SPZ für chronisch kranke Kinder Leitung, Abt. Interdisziplinär Pädiatrische Endokrinologie und Diabetologie Augustenburger Platz 1, (Mittelallee 7a) D 13353 Berlin E-Mail: dirk.schnabel@charite.de |
Prof. Dr. med. Dieter Haffner Klinik für Pädiatrische Nieren-, Leber- und Stoffwechselerkrankungen und Neuropädiatrie Zentrum für Kinderheilkunde und Jugendmedizin Medizinische Hochschule Hannover Carl-Neuberg Str.1 D 306325 Hannover E-Mail: haffner.dieter@mh-hannover.de |