
What is phosphate diabetes(XLH)?
(Deutsche Version)
X-linked dominant hypophosphatemic rickets (XLH) is a congenital disorder that affects not only the skeletal system but also the mineralization of teeth.
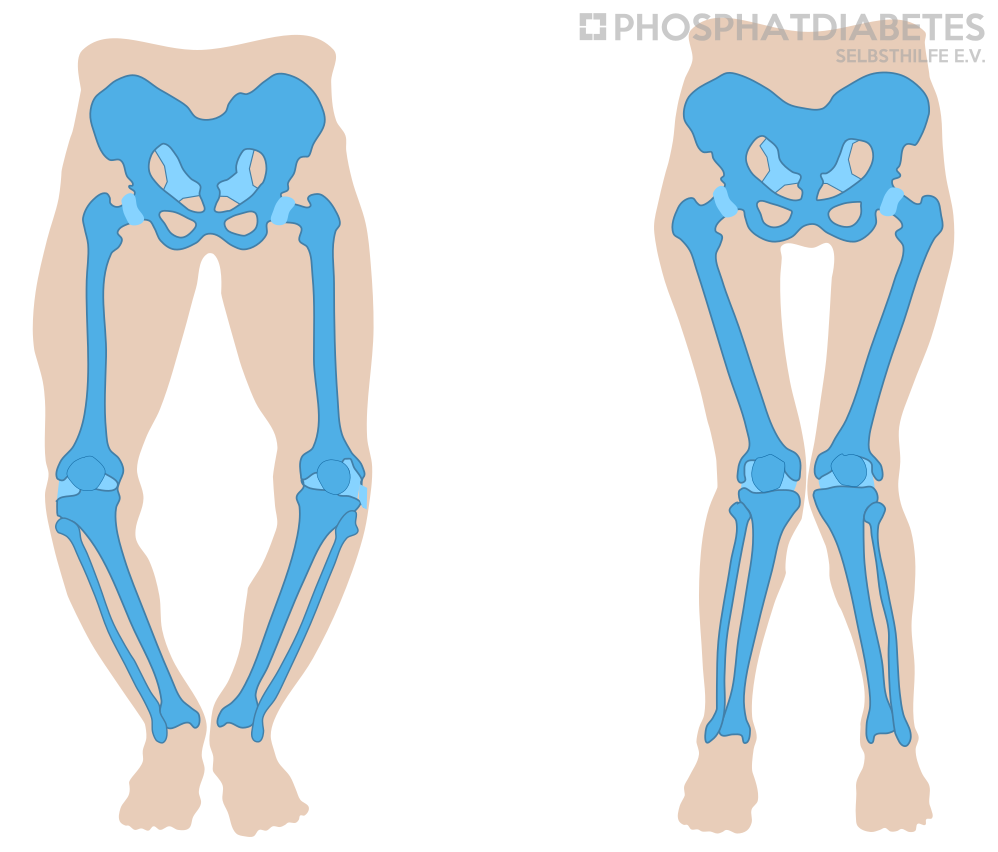
The illness can be noticed from the end of the first year of life, usually with the age to learn walking in the second year of life, by a waddling, broad-legged gait, and a decreased length growth. In some patients the diagnosis can be delayed until school age due to leg pain and/or increasing leg axis deformities (bowlegs or knock-knees).
Cause of the disease
A healthy, solid bone consists of a basic bone substance and a specific balance of the mineral’s calcium and phosphate. If one of these three important components are out of balance, the bone may become soft and, especially in the lower extremities, the lower legs and/or upper legs may bow or even fracture.
In the above-mentioned form of hypophosphatemic rickets (XLH), a genetic change in the so called PHEX gene leads to increased production of the hormone fibroblast growth factor 23 (FGF23). FGF23 is produced in the bone cells and normally regulates the phosphate balance of the body in addition to vitamin D. However, if the genetic mutation leads to an expanded formation of FGF23, it results to an increased excretion of phosphate via the kidneys. Normally 90% of the phosphate filtered by the kidneys is returned to the body. However, the heightened concentration of FGF23 causes a dysfunction of this reabsorption and to a disproportionately high excretion of phosphate via the kidneys into the urine. This initiates a low phosphate level in the blood and thus a reduced concentration of phosphate in the skeletal system. In addition, FGF23 decreases the synthesis of active vitamin D in the kidneys. The phosphate deficiency in the bones and a lack of active vitamin D results in a mineralization disorder and thus in a softness of the bones, which is particularly evident in the legs and is known as hypophosphatemic (= deficient phosphate) rickets.
The disease is linked to the X chromosome. This means that if the father has the disease, all daughters are affected and all sons are healthy, while statistically half of the sons and half of the daughters of a mother with XLH can be affected. The severity of the disease is quite different even within a family. Meanwhile, more than 300 different disease-triggering genetic changes in the PHEX gene (mutations) have been described. In addition to the inherited form of hypophosphatemia, there are patients from families in which neither father nor mother previously had XLH. This is the case in about one third of patients. This is known as a spontaneous mutation.
The incidence of this rare bone disease is estimated to be 20000 to 25000 new-borns per affected new-born. Despite this rareness, it is the most common congenital form of rickets.
Diagnosis
If, after the start of free walking in the second year of life, there is an increasing bowing of the lower legs (O-legs) or if a child does not like to walk at the end of the second year of life, the paediatrician should perform a blood test. In this examination, a vitamin D deficiency must first be ruled out. This is much more common than XLH and can also be the cause of leg deformities in young children. However, if the parents have carried out vitamin D prophylaxis in the first and second years of life, a vitamin D deficiency as a cause of bowed legs is very unlikely. If the blood test shows a reduced serum phosphate value (<1 mmol/l) and the marker for bone turnover is increased (alkaline phosphatase), the child should be seen by a specialist for hormone disorders (paediatric endocrinologist: www.dgked.de) or by a specialist for kidney diseases (paediatric nephrologist: www.gpn.de).
These specialists then perform further laboratory tests to confirm or invalidate the suspected diagnosis of XLH. In addition to supplementary laboratory values, an X-ray of the left hand is usually required. In this x-ray, rachitic changes could then be found in case of XLH. These are indications of a disordered mineralization of the bone. The disease can be further confirmed by a genetic examination in about 80% of patients. This should be done if the family history is negative or the findings are unclear.
In children affected by the disease, the diagnosis can be made in the first weeks of life before symptoms manifest themselves, either by laboratory chemical analysis and/or, if a mutation is known, by genetic testing.
Therapy
Treatment for this form of hypophosphatemic rickets is either with „conventional“ drug therapy consisting of at least 4 to 6 doses of phosphate distributed over the day (mostly as a liquid, in older children also by taking phosphate capsules) in combination with active vitamin D (active ingredient: Alfacalcidol or Calcitriol) as liquid or tablets. Alternatively, there is an antibody therapy, which is given from the age of 2 years with the registered drug Burosumab (see below).
With the combination of active vitamin D and phosphate, an attempt is made to absorb more of the phosphate supplied through food via the intestines and to pass it on to the body.
Ideally, it is possible to transport enough phosphate into the skeletal system that the activity of the bone turnover value (alkaline phosphatase in serum) is almost normalized, the bone is well mineralized and thus becomes stable and resilient and growth improves. The earlier the therapy is initiated, the lower the risk of developing severe leg axis deformities.
Unfortunately, problems can occur when taking phosphate, because some children do not like the taste of phosphate. Larger doses of phosphate can lead to abdominal pain and diarrhoea. It is therefore important to divide the daily phosphate amount into as many small doses as possible.
Since spring 2018, an approved therapy with an antibody (Burosumab), which specifically binds and neutralizes the elevated endogenous FGF23, has been available for XLH patients from the age of 2 years with the radiological confirmation of a bone disease. The drug containing the active ingredient Burosumab is subcutaneous injected every 14 days. This is carried out professionally by a homecare service.
Under the therapy, the originally significantly increased phosphate excretion via the urine usually returns to normal after a few injections. The blood phosphate level rises to the lower normal value range. The skeletal system is now sufficiently supplied with phosphate, and skeletal mineralization is increasingly improving. In the medium to long term, X-rays show a significant improvement in bone mineralization. After several years of therapy, the leg axis deformation has also been observed to have become straightening. However, this can also be observed under conventional therapy.
After the start of therapy, initially in the first few weeks, short-term appointments are required to check tolerance, but also to adjust the dose of Burosumab. This is controlled by blood and urine tests. It is not uncommon for an increase in the drug dose to be necessary.
The medication is usually well tolerated, severe side effects have not been reported so far. There are no findings about possible long-term side effects, which must be monitored further.
The pros and cons of the two therapeutic options have been described in detail in a European therapy recommendation (see below).
Control of drug adjustment
After the beginning of the drug therapy, whether using phosphate and vitamin D or through injection of the antibody, it is recommended that the child or adolescent is seen by a specialist every 4 to 6 weeks, later every 3 months. During these check-ups, the child’s urine and blood are analysed. Depending on the laboratory values, the specialist may then adjust the previous medication.
Recommended additional examinations
Kidneys
Drug therapy with phosphate and active vitamin D can sometimes lead to calcium deposits in the kidneys (nephrocalcinosis). Therefore, an ultrasound examination of the kidneys should be performed about every one to two years.
Skeletal system
In patients with XLH, mostly the upper and/or lower legs are often bent in a strong X or O position. This results in a conspicuous gait pattern. The earlier the patient is diagnosed, and treatment is started, the greater the possibility that this leg deformity will correct itself again under sole medication. However, if this does not occur after a consistent drug therapy over 3 to 4 years, growth control by using epiphysiodesis (e.g., so-called eight-plates) should be considered. In older children / adolescents, a major surgical intervention („repositioning osteotomy“) may also be necessary.
Growth
Children with XLH are usually of normal size and weight at birth. From the age of 2 years on, a slower growth rate results in a worse body length growth than one would expect with the size of the parents. Even with good medication and straight legs, patients with XLH as adults are usually only at the border of normal growth but can be quite normal as adults with large parents.
Teeth
Patients with XLH may also have a disorder of enamel and dentine development of milk teeth, but also of permanent teeth. Fistulas, inflammatory changes of the tooth root tips with suppurations (abscesses) are often seen in caries-free dentition. To prevent possible tooth loss, regular dental examinations are recommended. First and foremost, however, proper daily dental care and regular prophylaxis procedures are essential.
Hearing
Very rarely, a hearing disorder in the inner ear occurs in the course of the disease. If there are indications of a hearing disorder, it is recommended to consult an ear, nose and throat specialist.
Development of the head
Very rarely, a conspicuous development of the skull (craniosynostosis) occurs in infancy and toddlerhood. In such cases, the paediatrician should be contacted for a consultation with the paediatric neurosurgeon.
Outlook to the future
Extension of the treatment age for the Burosumab therapy: In 2020 a phase 2 study for the treatment with Burosumab from infancy to adolescence for babies was started in which 3 German centres are also participating.
On October 2, 2020, the European Commission granted an extension of approval for the treatment of older adolescents and adults with X-linked hypophosphatemic rickets (XLH).
As a result, this treatment is now also available for adolescents with radiological evidence of bone disease, regardless of growth status.
European clinical practice recommendation for patients with XLH
In December 2017, a group of 20 experts from the fields of paediatrics, nephrology, endocrinology, orthopedy, dentistry and neurosurgery, as well as representatives of the patient organization Phosphatdiabetes e.V. from Germany and the patient organization from France met in Berlin to develop therapy recommendations for this rare disease. These recommendations have been published in a very renowned journal since July 2019 (Haffner et al Nat Rev Nephrol 2019) and can be viewed free of charge as an open access publication. A German translation is currently being worked on.
Patient registry
To learn more about the course of the disease and the effectiveness of XLH therapy in children and adolescents, paediatric endocrinologists and paediatric nephrologists from Germany have joined to initiate a patient registry. In this process, the relevant clinical data of the patients will be transferred to a database after the parents and patients have been informed and given their written agreement and the personal data have been made unrecognizable („pseudonymization“). In the meantime, 20 HCPs with more than 50 patients have already been included in the register. The inclusion of 150 patients from 39 centres is planned. The participants of the study registry meet once a year. Further clinical trials for the treatment of the disease are also planned within the registry.
In addition, registries for adult XLH patients will be initiated, e.g., via the ERNs (European Reference Networks).
Summary
XLH is a rare congenital disease that mainly affects the skeletal system and teeth of patients. Early diagnosis and treatment are necessary to prevent complications, especially of the skeletal system but also of other organs. With the development and approval of Burosumab, the possibilities of drug therapy have been significantly expanded. For the first time, normal phosphate concentrations in the blood of the treated patients can be detected, leading to a significant improvement in bone mineralization after approximately 9 to 12 months. Due to the involvement of different organs in the disease, care should be provided by a multidisciplinary team in a special consultation hour if possible.
This document was translated from German to English by Martha Kirchhoff, a medical layman.
Literature
Haffner D, Waldegger S (2017). Disorders of phosphorus metabolism. In: Pediatric Kidney Disease. 1. Edition. Editors: D.-F. Geary, F. Schaefer. Springer, Heidelberg. S. 953-972
Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, Kamenicky P, Nevoux J, Prié D, Rothenbuhler A, Wicart P, Harvengt P. Therpeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 2014 Mar 14;3(1):R13-30
Schnabel D (2014). Störungen des Kalzium-Phosphat-Stoffwechsels, in: Hoffmann GF, Lentze MJ, Spranger J, Zepp F (Ed.). Pädiatrie – Grundlagen und Praxis, 4.Auflage: 612-631, Band 1. Springer Verlag, Heidelberg
| Dr. med. Dirk Schnabel, Ltd. Oberarzt Stellv. Leiter SPZ für chronisch kranke Kinder Leitung, Abt. Interdisziplinär Pediatric Endocrinology and Diabetology Augustenburger Platz 1, (Mittelallee 7a) D 13353 Berlin E-Mail: dirk.schnabel@charite.de |
Prof. Dr. med. Dieter Haffner Clinic for Pediatric Kidney, Liver and Metabolic Diseases Zentrum für Kinderheilkunde und Jugendmedizin Medizinische Hochschule Hannover Carl-Neuberg Str.1 D 306325 Hannover E-Mail: haffner.dieter@mh-hannover.de |